OpRegen®
for Dry AMD

OpRegen® (RG6501) is an allogeneic retinal pigmented epithelial (RPE) cell replacement therapy currently in Phase 2a development under a worldwide collaboration with Roche and Genentech for the treatment of geographic atrophy (GA) secondary to dry-AMD.
Our approach is to replace the layer of damaged RPE cells with new, healthy, and functional RPE cells manufactured from a well-characterized, allogeneic cell line, transplanted to the subretinal space, and extensively cover the area of geographic atrophy.
The goal of the OpRegen therapeutic approach is to slow or halt disease progression and to preserve and/or restore visual function in patients affected by dry-AMD. OpRegen has been granted Fast Track and Regenerative Medicine Advanced Therapy (RMAT) designations from the FDA, which includes an expedited regulatory path with the ability for increased interfacing with the FDA during the clinical development process.

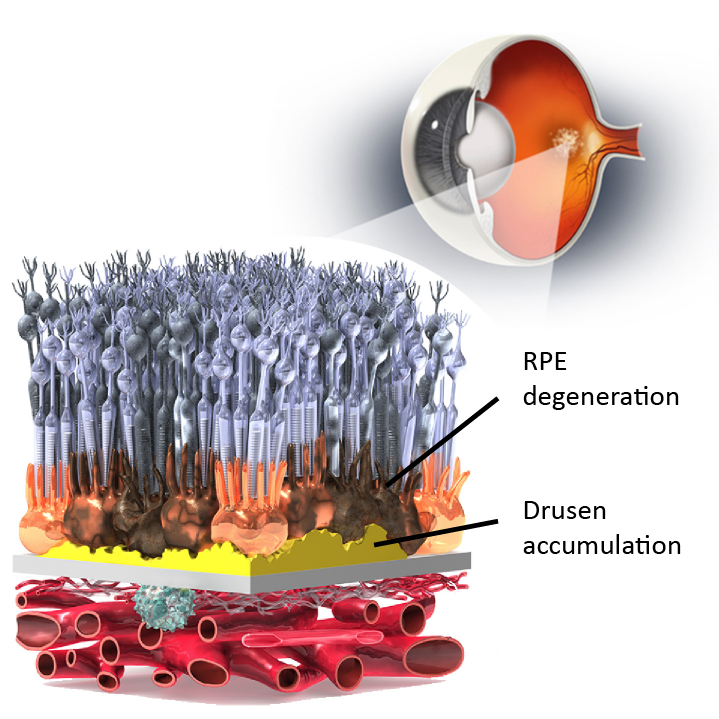
Image adapted from scienceofamd.org
OpRegen is currently being evaluated in a Phase 2a study intended to optimize subretinal surgical delivery and evaluate safety and activity in up to 60 patients. The primary objectives of the study are to evaluate (i) the proportion of patients with subretinal surgical delivery of OpRegen to target regions under the retina, and (ii) to evaluate the safety of subretinal surgical delivery of OpRegen as measured by the incidence and severity of procedure-related adverse events at 3 months following surgery. A key secondary objective is to evaluate the proportion of patients with qualitative improvement in retinal structure, as determined by Optical Coherence Tomography (SD-OCT) imaging, within 3 months following surgery.
In June 2025, 36-month visual acuity results from patients enrolled in a Phase 1/2a clinical study of OpRegen in patients with GA secondary to AMD, were presented at the Clinical Trials at the Summit 2025.
Clinical Trials at the Summit 2025 Highlights
The Clinical Trials at the Summit 2025 presentation is available on the Events and Presentations section of Lineage’s website.
BCVA gains in study eyes are sustained
in Cohort 4 (Less advanced GA) patients 36 Months Post-Treatment and BCVA Gains are Greater Among Eyes with Extensive Coverage of GA by the Surgical Bleb

Presented at the Clinical Trials at the Summit 2025
Sustained Evidence of Retinal Structural Support
in Cohort 4 (Less advanced GA) patients 36 Months Post-Treatment by Quantitative OCT Analysis

Sustained Evidence of Retinal Structural Support is Most Evident Among Eyes with Extensive Coverage of GA by the Surgical Bleb

Presented at the Clinical Trials at the Summit 2025
GAllete Study Design
A Phase 2a Multicenter, Open-Label, Single-Arm Study to Optimize the Subretinal Delivery of Opregen Cell Therapy In GA (NCT05626114)

Presented at the Clinical Trials at the Summit 2025
Advanced Subretinal Delivery Devices
Under Development for Evaluation with OpRegen Cell Therapy

Presented at the Clinical Trials at the Summit 2025